| PLS_Toolbox Documentation: alignmat | < abline | als > |
alignmat
Purpose
Alignment of matrices and N-way arrays
Synopsis
[bi,itst] = alignmat(amodel,b);
[bi,itst] = alignmat(a,b,nocomp);
Description
In some cases, data arrays require alignment to aid the performance of the three-way (e.g. GRAM, or PARAFAC) or unfold models such as MPCA. For example, sometimes GC peaks or data from batch operations can be shifted on a sample-to-sample basis (each sample is a Mb by N matrix). In these cases, it is advantageous to choose a sub-matrix of a single matrix A as a standard and find the sub-matrix of subsequent samples B that best align or match the standard matrix. It is also possible to use a model of one or more standard matrices Amodel and find the sub-matrix of subsequent samples B that best align or match the model. In the latter case, it is also possible to find the sub-array of B that best aligns with the model of a N-way data set (Amodel). This can be performed along multiple modes using ALIGNMAT.
ALIGNMAT finds the subarray of b, bi, that most matches a using two different algorithms. For input:
[bi,itst] = alignmat(amodel,b);
the sub-array bi is found using a projection method. In this case, bi is the sub-array of b that has the lowest residuals on a model of a called amodel. Models for amodel are standard model structures from PCA, PCR, GRAM, TLD, or PARAFAC. Input b can be class "double" or "dataset" and must have the same number of modes/dimensions as a with each element of size(b) ≥ size(a). Alignment is performed for modes with size(b) > size(a).
For input:
[bi,itst] = alignmat(a,b,ncomp);
both a and b can be class "double" or "dataset", but both are two-way arrays (matrices). For a M by N then b must be Mb by N where Mb ≥ M (when Mb = M no alignment is performed). The output bi is the sub-array of b that best matches the matrix a. Optional input ncomp is a scalar of the number of components to use in the decomposition {default: ncomp = 1}.
Output bi is an array of class "double", itst is a cell array containing the indices of b that match bi. Note that since interpolation is used the indices in itst are not in general integers.
Algorithm
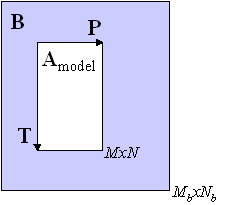 For the projection method, Amodel is a
model of array A. This can be a model from PCA, GRAM, TLD, or PARAFAC.
For example, if A is a M by N matrix then the PCA model of
A is
For the projection method, Amodel is a
model of array A. This can be a model from PCA, GRAM, TLD, or PARAFAC.
For example, if A is a M by N matrix then the PCA model of
A is ![]() where
T is M by K and P is N by K. Alignmat
finds the submatrix of B, Bi, that has the lowest
residuals on the model of A i.e.
where
T is M by K and P is N by K. Alignmat
finds the submatrix of B, Bi, that has the lowest
residuals on the model of A i.e.  . This can be used to find the data
“cube” within N-way arrays.
. This can be used to find the data
“cube” within N-way arrays.
In the figure, this is represented as having each of the M by N sub-matrices of B projected onto the model of the M by N model of A. Note that in the figure that the size of B is Mb by Nb with Mb>M and Nb>N.
The projection method was presented in Gallagher, N.B. and Wise, B.M., “Standardization for Three-Way Analysis”, TRICAP 2000: Three-way Methods in Chemistry and Psychology, Hvedholm Castle, Faaborg, Denmark, July (2000). In that study, it was found that the projection method was faster and more robust than the SVD-based algorithm discussed below.
 In the SVD method,
the standard matrix A and a sub-matrix of B, Bi,
are aumented and a singular value decomposition of the result is performed such
that [u,s,v] = svd([AMxN|BiMxNb]).
The sub-matrix is incremented and the SVD is performed again. The sub-matrix
that minimizes the rank is selected as matching best. The objective function is
In the SVD method,
the standard matrix A and a sub-matrix of B, Bi,
are aumented and a singular value decomposition of the result is performed such
that [u,s,v] = svd([AMxN|BiMxNb]).
The sub-matrix is incremented and the SVD is performed again. The sub-matrix
that minimizes the rank is selected as matching best. The objective function is
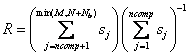 .
Note that in this algorithm N and Nb do not have to be
equal. The algorithm is discussed in Prazen, et al., Anal. Chem.,
70, 218-225, 1998.
.
Note that in this algorithm N and Nb do not have to be
equal. The algorithm is discussed in Prazen, et al., Anal. Chem.,
70, 218-225, 1998.
See Also
analysis, gram, parafac, pca, registerspec, tld
| < abline | als > |